Making Genomic Data Analysis Faster and More Accurate
The Human Genome Project was completed almost a decade ago. One of its most promising potential outcomes is the ability to use genomic data for personalized medicine, especially in genetically-driven diseases such as cancer. Despite the excitement around this approach, personalized medicine is still in its infancy. One hurdle is producing large amounts of low-cost but accurate genomic data, to better understand the connections between genotype and health. Fortunately, technology advances in the past ten years have enabled a rapid fall, faster than Moore’s Law, in the cost of DNA sequencing. However, the disadvantage of new DNA sequencers is that postprocessing the data they produce is hard. These sequencers produce large numbers of short (100-character) “reads” from the genome, which must then be assembled, much like a puzzle, into a full sequence. Because of its high computational cost, this data analysis problem will soon dominate the cost of reconstructing a genome.
Our group, a team of researchers from the UC Berkeley AMP Lab, Microsoft, and UCSF, is working on a holistic system to quickly and accurately process short-read DNA data. This is in contrast to previous work, where the processing was broken up into discrete stages and separately optimized, resulting in inefficient resource usage and information loss. Our most mature contribution so far is a new algorithm for the first step in this process, alignment, where each read is matched to the location in the genome from which it most likely came. Alignment has traditionally been highly compute-intensive, taking days for one genome. Our new aligner, the Scalable Nucleotide Alignment Program (SNAP), reduces this cost by 10-100x, while simultaneously improving accuracy. It accomplishes this through a combination of algorithmic innovation and judicious use of modern hardware. We are also applying the insights from SNAP to further steps of the sequencing process, which use alignment results from multiple reads to determine the individual’s true genotype.
Speaker Details
Matei Zaharia is a fifth-year PhD student at UC Berkeley, working with Scott Shenker and Ion Stoica on topics in computer systems, networks and cloud computing. He is also a committer on Apache Hadoop and Apache Mesos. Matei is funded by a Google PhD Fellowship.
Kristal Curtis is a fifth-year PhD student in the AMP Lab at UC Berkeley, advised by David Patterson and Armando Fox. Her research has focused on performance modeling for storage systems and fast and accurate analysis of genomics data. She has been supported by an NSF Graduate Research Fellowship and a UC Berkeley Chancellor’s Fellowship.
- Series:
- Microsoft Research Talks
- Date:
- Speakers:
- Matei Zaharia and Kristal Curtis
- Affiliation:
- UC Berkeley
-
-

Jeff Running
-
-
Series: Microsoft Research Talks
-
-
-
-

Galea: The Bridge Between Mixed Reality and Neurotechnology
Speakers:- Eva Esteban,
- Conor Russomanno
-

Current and Future Application of BCIs
Speakers:- Christoph Guger
-

Challenges in Evolving a Successful Database Product (SQL Server) to a Cloud Service (SQL Azure)
Speakers:- Hanuma Kodavalla,
- Phil Bernstein
-

Improving text prediction accuracy using neurophysiology
Speakers:- Sophia Mehdizadeh
-
-

DIABLo: a Deep Individual-Agnostic Binaural Localizer
Speakers:- Shoken Kaneko
-
-

Recent Efforts Towards Efficient And Scalable Neural Waveform Coding
Speakers:- Kai Zhen
-
-
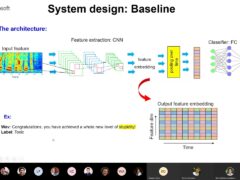
Audio-based Toxic Language Detection
Speakers:- Midia Yousefi
-
-
From SqueezeNet to SqueezeBERT: Developing Efficient Deep Neural Networks
Speakers:- Sujeeth Bharadwaj
-
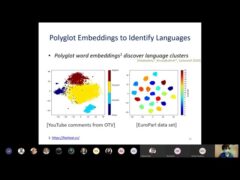
Hope Speech and Help Speech: Surfacing Positivity Amidst Hate
Speakers:- Monojit Choudhury
-
-
-
-
-

'F' to 'A' on the N.Y. Regents Science Exams: An Overview of the Aristo Project
Speakers:- Peter Clark
-

Checkpointing the Un-checkpointable: the Split-Process Approach for MPI and Formal Verification
Speakers:- Gene Cooperman
-

Learning Structured Models for Safe Robot Control
Speakers:- Ashish Kapoor
-
-