Quantum Computation for Quantum Chemistry: Status, Challenges, and Prospects – Session 3
1:15 – 2:00PM
Challenges of Electronic Structure Calculations on Quantum Computers
Speaker: Sabre Kais, Purdue University and Qatar Environment and Energy Research Institute
Abstract:
The exact electronic structure calculations of atoms and molecules on classical computers generally scale exponentially with the size of the system. Using quantum computers, the computational resources required to carry out the simulation are polynomial. I will present three related approaches to electronic structure: The quantum circuit model, the variational model and the adiabatic quantum computing model and discuss the opportunities, open questions and challenges in this field.
Bio:
In 1989, Prof. Kais received his Ph.D. in Chemical Physics from the Hebrew University. After a postdoctoral appointment at Harvard University 1989-1994, he joined the Chemistry Department at Purdue in 1994 as an Assistant Professor. Since 2002, he has been a full professor of Chemical Physics. He received the National Science Foundation Career Award in 1997. He was also awarded the Purdue University Faculty Scholar Award (2004-2009), and the Guggenheim Fellowship Award (2005). In 2007, Prof. Kais was the Elected Fellow of the American Physical Society and the Elected Fellow of the American Association for the Advancement of Science. In 2012, he received the Sigma Xi Research Award. He has courtesy professorship appointments at both the Department of Computer Science and the Department of Physics at Purdue and member of Qatar Environment and Energy Research Institute. He has published over 140 peer-reviewed papers and served on the editorial board of several chemistry and physics journals. Recently, with his colleagues, established a new center, Quantum Information for Quantum Chemistry (QIQC) http://web.ics.purdue.edu/~kais/qc/
2:00 – 2:45PM
Fermionic Quantum Simulation: From Jordan-Wigner to Bravyi-Kitaev
Speaker: Peter J Love, Haverford College
Abstract:
Simulation of fermionic systems has been a topic of interest in quantum simulation since
Feynman’s first papers on the topic. It has been known for some time how to simulate fermionic
systems and scalable proposals for electronic structure calculations on quantum computers require some solution to this problem. Current work makes use of the Jordan-Wigner transformation to track phases arising from exchange anti-symmetry. For a single term in a fermionic Hamiltonian on N modes the Jordan wigner transformation requires an overhead of O(N) gates. In this talk I will give an alternative to the Jordan Wigner transformation, originally developed by Bravyi and Kitaev, which reduces this overhead to O(log N). We give the details of this transformation for electronic structure Hamiltonians and give the minimal basis model of the Hydrogen molecule as an example.
Bio:
Professor Love completed his undergraduate degree in Physics at Oxford University in 1997, during which he was awarded summer internships at NASA Goddard space flight centre and at the Joint European Torus. He completed a D.Phil in Theoretical Physics at Oxford University under the supervision of Dr. J. M. Yeomans in 2001, specialising in lattice-gas cellular automata models of complex fluids. He then completed a 1 year Post-Doctoral appointment with Prof. P. V. Coveney at the Centre for Computational Science, Department of Chemistry, Queen Mary, University of London, continuing his work on LGCA’s and co-authoring a £3.3 million Grid based computing proposal funded by the EPSRC. He then completed a DARPA funded post-doctoral position at Tufts University Department of Mathematics, after which he moved to D Wave Systems in November 2004 as Senior Applications Scientist. In July 2006 he took a position as an Assistant Professor of Physics at Haverford College, where he was the recipient of the 2009 Christian and Mary Lindback foundation award for distinguished teaching and was promoted to Associate Professor in 2012. Prof. Love is the PI a 2009 NSF Career Award and the co-PI of the NSF Quantum Information for Quantum Chemistry Center for Chemical Innovation.
2:45 – 3:30PM
Error Correction and Architectures for the Simulation of Quantum Materials on a Quantum Computer
Speaker: Ken Brown, Georgia Tech
Abstract: Quantum computers promise algorithmically faster calculations of molecular properties by performing operations on the whole quantum mechanical state space. The challenge of implementing these algorithms is the development of reliable quantum hardware. In principle, this hardware problem can be solved by fault-tolerant quantum error-correction. Fault-tolerant quantum error-correction comes with additional requirements that affect the total computational resources necessary to calculate the molecular properties. In this talk, we will examine this additional resource cost and propose theoretical and experimental targets for reducing the resource cost.
Bio:
Kenneth Brown received his B.S. in Chemistry from the University of Puget Sound in 1998. For his PhD, he studied theoretical quantum information science as a Hertz Fellow at UC Berkeley. Following his PhD, he held a postdoctoral position at MIT where he performed quantum information experiments using NMR and ion traps. Since 2007, he has been an assistant professor at Georgia Tech where he leads a research group studying quantum information and cold molecular ions.
Speaker Details
In 1989, Prof. Kais received his Ph.D. in Chemical Physics from the Hebrew University. After a postdoctoral appointment at Harvard University 1989-1994, he joined the Chemistry Department at Purdue in 1994 as an Assistant Professor. Since 2002, he has been a full professor of Chemical Physics. He received the National Science Foundation Career Award in 1997. He was also awarded the Purdue University Faculty Scholar Award (2004-2009), and the Guggenheim Fellowship Award (2005). In 2007, Prof. Kais was the Elected Fellow of the American Physical Society and the Elected Fellow of the American Association for the Advancement of Science. In 2012, he received the Sigma Xi Research Award. He has courtesy professorship appointments at both the Department of Computer Science and the Department of Physics at Purdue and member of Qatar Environment and Energy Research Institute. He has published over 140 peer-reviewed papers and served on the editorial board of several chemistry and physics journals. Recently, with his colleagues, established a new center, Quantum Information for Quantum Chemistry (QIQC) http://web.ics.purdue.edu/~kais/qc/
Professor Love completed his undergraduate degree in Physics at Oxford University in 1997, during which he was awarded summer internships at NASA Goddard space flight centre and at the Joint European Torus. He completed a D.Phil in Theoretical Physics at Oxford University under the supervision of Dr. J. M. Yeomans in 2001, specialising in lattice-gas cellular automata models of complex fluids. He then completed a 1 year Post-Doctoral appointment with Prof. P. V. Coveney at the Centre for Computational Science, Department of Chemistry, Queen Mary, University of London, continuing his work on LGCA’s and co-authoring a £3.3 million Grid based computing proposal funded by the EPSRC. He then completed a DARPA funded post-doctoral position at Tufts University Department of Mathematics, after which he moved to D Wave Systems in November 2004 as Senior Applications Scientist. In July 2006 he took a position as an Assistant Professor of Physics at Haverford College, where he was the recipient of the 2009 Christian and Mary Lindback foundation award for distinguished teaching and was promoted to Associate Professor in 2012. Prof. Love is the PI a 2009 NSF Career Award and the co-PI of the NSF Quantum Information for Quantum Chemistry Center for Chemical Innovation.
Kenneth Brown received his B.S. in Chemistry from the University of Puget Sound in 1998. For his PhD, he studied theoretical quantum information science as a Hertz Fellow at UC Berkeley. Following his PhD, he held a postdoctoral position at MIT where he performed quantum information experiments using NMR and ion traps. Since 2007, he has been an assistant professor at Georgia Tech where he leads a research group studying quantum information and cold molecular ions.
- Series:
- Microsoft Research Talks
- Date:
- Speakers:
- Sabre Kais, Peter J Love, and Ken Brown
- Affiliation:
- Purdue University, Haverford College, Georgia Tech
-
-

Jeff Running
-

Krysta M. Svore
General Manager
-
Series: Microsoft Research Talks
-
-
-
-

Galea: The Bridge Between Mixed Reality and Neurotechnology
Speakers:- Eva Esteban,
- Conor Russomanno
-

Current and Future Application of BCIs
Speakers:- Christoph Guger
-

Challenges in Evolving a Successful Database Product (SQL Server) to a Cloud Service (SQL Azure)
Speakers:- Hanuma Kodavalla,
- Phil Bernstein
-

Improving text prediction accuracy using neurophysiology
Speakers:- Sophia Mehdizadeh
-
-

DIABLo: a Deep Individual-Agnostic Binaural Localizer
Speakers:- Shoken Kaneko
-
-

Recent Efforts Towards Efficient And Scalable Neural Waveform Coding
Speakers:- Kai Zhen
-
-
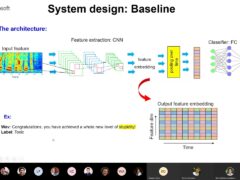
Audio-based Toxic Language Detection
Speakers:- Midia Yousefi
-
-
From SqueezeNet to SqueezeBERT: Developing Efficient Deep Neural Networks
Speakers:- Sujeeth Bharadwaj
-
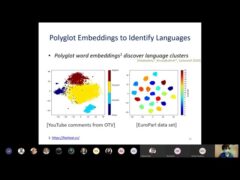
Hope Speech and Help Speech: Surfacing Positivity Amidst Hate
Speakers:- Monojit Choudhury
-
-
-
-
-

'F' to 'A' on the N.Y. Regents Science Exams: An Overview of the Aristo Project
Speakers:- Peter Clark
-

Checkpointing the Un-checkpointable: the Split-Process Approach for MPI and Formal Verification
Speakers:- Gene Cooperman
-

Learning Structured Models for Safe Robot Control
Speakers:- Ashish Kapoor
-
-